The intrinsic parameters of carbon nanotubes (CNTs) such as ionization potential (IP) and electron affinity (EA) are closely related to their unique properties and associated applications. In this work, we demonstrated the success of optimal tuning method based on rangeseparated (RS) density functionals for both accurate and efficient prediction of vertical IPs and electron affinities (EAs) of a series of armchair single-walled carbon nanotubes C20nH20 (n = 2–6) compared to the high-level IP/EA equation-of-motion coupled-cluster method with single and double substitutions (IP/EA-EOM-CCSD). Notably, the resulting frontier orbital energies (–εHOMO and –εLUMO) from the tuning method exhibit an excellent approximation to the corresponding IPs and EAs, that significantly outperform other conventional density functionals. In addition, it is suggested that the RS density functionals that possess both a fixed amount of exact exchange in the short-range and a correct long-range asymptotic behavior are suitable for calculating electronic structures of finite-sized CNTs. Next the performance of density functionals for description of various molecular properties such as chemical potential, hardness and electrophilicity are assessed as a function of tube length. Thanks to the efficiency and accuracy of this tuning method, the related behaviors of much longer armchair single-walled CNTs until C200H20 were studied. Lastly, the present work is proved to provide an efficient theoretical tool for future materials design and reliable characterization of other interesting properties of CNT-based systems.
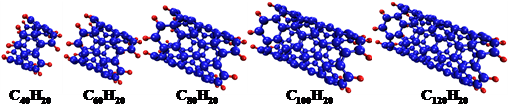
图1 不同长度的单壁碳纳米管结构示意图
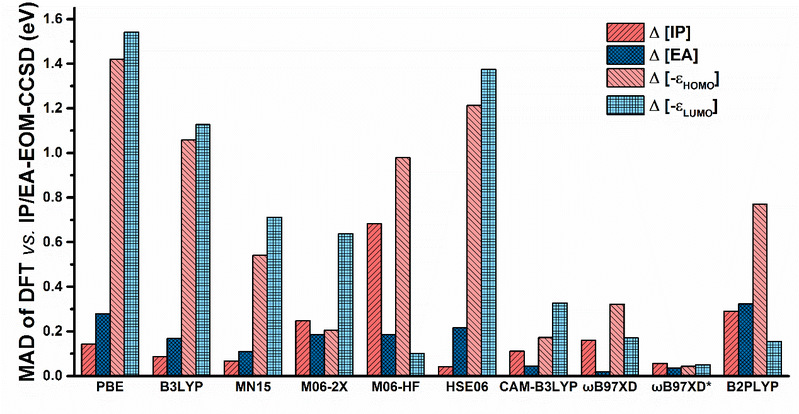
图2 各种密度泛函理论方法计算能级参数的误差分布柱形图
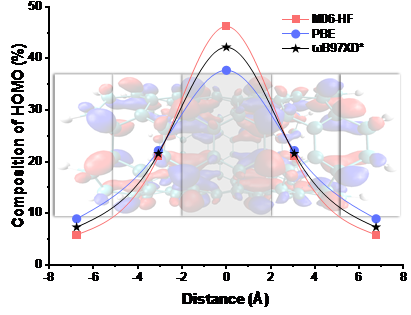
图3 各片段对不同泛函方法优化的C120H20结构下的HOMO轨道的贡献图
